What is DIA Proteomics?
Classic LC-MS/MS-based bottom-up proteomics separate peptides based on three dimensions: chromatographic retention time (RT); mass-to-charge ratio (m/z) and ion intensity.
Ion mobility analysis is a gas-phase technique allowing the separation of ions based on their mobility through an inert gas (typically helium or nitrogen) under the influence of an electric field. Adding ion mobility as an additional dimension of separation for peptide ions to LC-MS/MS analysis make MS-based proteomics enter a new era.
It significantly improves the scanning speed and detection sensitivity, as well as enhances proteomic analysis performance in terms of identification depth, detection cycle, and quantitative accuracy.

Data Dependent/ Independent Acquisition (DDA/DIA)
There are two data acquisition strategies in tandem mass spectrometry (MS/MS) data acquisition:
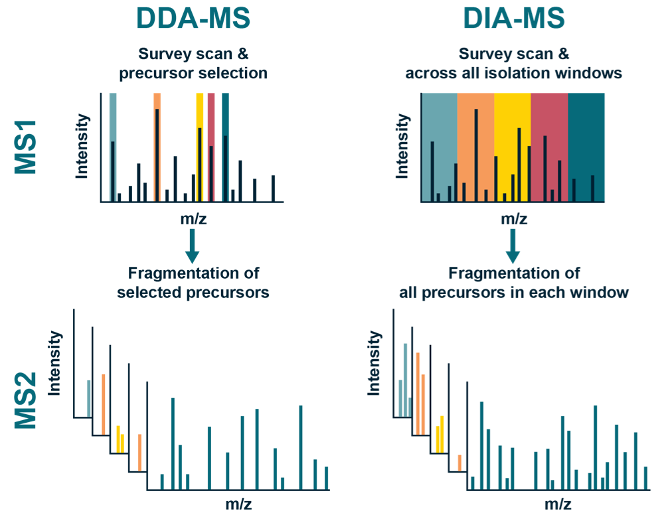
DDA mode: The mass spectrometer selects a fixed number of most intense precursor ions. They are then fragmented and analyzed, in the second stage of tandem mass spectrometry.
DIA mode: The mass spectrometer divides the full scan range of mass spectrometry into several windows and selects, fragments, and collects all ions in each window.
DIA strategy avoids data imbalance caused by randomness by realizing "lossless acquisition" of all possible data. However, the "all-acquisition" strategy produces a highly complex spectrogram that poses a great challenge to data analysis.
Technical Strengths of DIA Quantitative Proteomics
In the 4D-diaPASEF scanning mode, the ion-mobility-related CCS value correlates well with m/z. This feature allows the Quadrupole to scan gradually to collect nearly 100% of the ion signals, greatly enhancing the sensitivity and depth of detection.
In comparison, in the traditional LC-MS/MS-based proteomics without ion mobility separation, only limited signals can be collected.
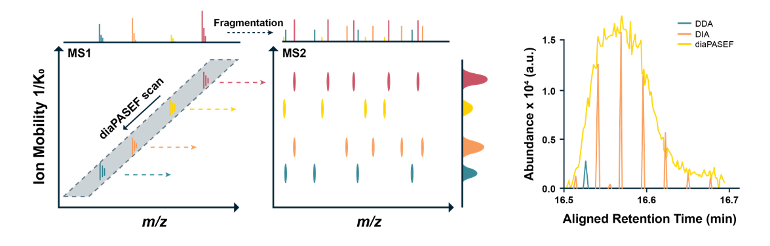
DIA identifies and quantifies low-abundant proteins more accurately than traditional methods, thereby increasing the depth of proteomics detection.
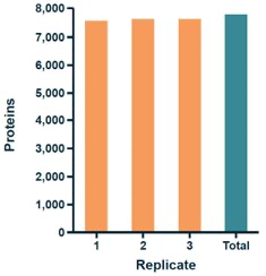
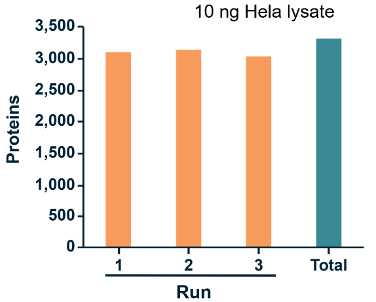
DIA technology can further push the limits of sensitivity with advanced instrument performance and upgraded acquisition methods. Even at this trace level, 4D-DIA showed 85% data integrity for protein quantification.